

Leiomyosarcoma Subtypes
Genetic lineages of leiomyosarcomas

Obligatory—This is not medical advice
For patients with sarcoma, the journey to find a physician who is familiar with their specific diagnosis can be a long one. Patients want to be seeing a doctor who has taken care of their disease before and has some base of knowledge and experience from which to draw. Their new physician can tell stories of other patients, treatments, and outcomes. The following discussion is an exploration of what had been seen in a group of patients with LMS, at a molecular level. Scientists are making observations through genetic profiling of leiomyosarcomas and subsequently relating those findings to outcomes. These are not perfect, and may not necessarily be applicable in many cases, but nonetheless, perhaps, help us think about how there may be genetic factors underlying the behavior of leiomyosarcomas.
Introduction
For more information about the basis of diagnosis and treatment of leiomyosarcomas, see some of my recent posts.
We know that LMS has very different phenotypes, despite carrying the same name. For instance, most sarcoma experts will relay information about cases of patients with LMS who have undergone metastasectomy (that is resection of metastatic leiomyosarcoma) and been cured. Meanwhile, there are likewise many patients who sadly are not so fortunate. While we may not always be able to understand what differentiates the biology of each individual’s tumor, a paper written in 2021 described, in general terms, three different patterns that were seen, and relate that broadly to survival and treatment.1
How did they do this?
The group took samples from patients at Mount Sinai Hospital and University Health Network in Toronoto, Canada. There were 113 patients evaluated, in total. Tumors from these patients had varying levels of genetic sequencing performed. The demographics were as to be expected for patients with LMS (average age of 56.5 years, female male ratio of 2.6:1).

They performed complex statistical analyses in order to cluster tumors based on specific genetic information. See the figure below. Cancers, of course, are not aware of such demarcation, and so you can see that there are clearly some tumors which fall between each delineation.

There were multiple other ways in which these genes bore relations to the anatomical and protein expression aspects of these tumors. All of these data appear to fit well into what we know about location and molecular characteristics of tumors.

What did it show?
In essence, this elaborate, and very detailed study demonstrated a potential future framework for genetic analysis for patients with leiomyosarcoma. The work here was exceptionally well done, but there will always be a need to prospectively validate such observations in trials. It is thought provoking and confirms the observation that in the cases of many cancers, the first step towards a clinically significant disease could begin years before diagnosis.2 ). Furthermore, there is in vitro evidence of the efficacy of PARP inhibition, which has been discussed elsewhere in this substack.
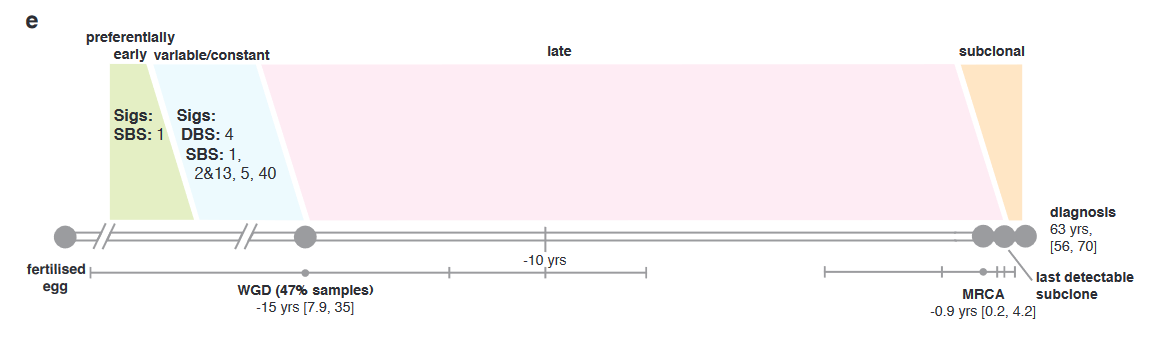
Small links between subtype (as defined by their analyses) and retrospective outcomes were made. Given that this, again, was something created by the authors, such links would need to be evaluated in the clinic prior to utilization in practice.

Conclusions
Leiomyosarcoma is a heterogeneous disease. The name leiomyosarcoma is a descriptor for what the pathologist can clearly see with their own eyes as manifest by various stains and shapes under the microscope. Clearly, patients and doctors are in need of more robust tools in order for more accurate prognosis and treatment recommendations. The paper above is largely descriptive, but nonetheless thought provoking. It confirms much of what has already been observed and fits nicely within the body of leiomyosarcoma literature. I would caution patients and providers from reading into this too deeply as, again, these observations only pertain to those individual samples and people studied, and not prospectively.
https://pubmed.ncbi.nlm.nih.gov/34301934/
https://www.nature.com/articles/s41586-019-1907-7