


Obligatory—This is note medical advice
Sometimes, patients are born with conditions that predispose them to developing different cancers. See a short, and very much incomplete table below. Oftentimes, patients have no idea until they’re told about their cancer diagnosis. These cases can be especially difficult because patients may be in the primes of their lives: teens, twenties, and early thirties (also known as adolescents and young adults or AYA). We counsel patients on their conditions, what we can do, and who else within their family should be tested. We also recommend genetic counseling with an expert in heritable syndromes.

As you can see in the table, neurofibromatosis type 1 is one of these conditions. We’ll discuss the pathogenesis of this condition, and Malignant Peripheral Nerve Sheath Tumors (MPNSTs) more, below.
How Common is Malignant Peripheral Nerve Sheath Tumor? What is it?
An MPNST is a soft tissue sarcoma (STS) which arises from peripheral nerve sheaths. MPNSTs can be aggressive tumors and are associated with frequent local recurrence and metastatic disease. Notably, half of MPNSTs arise in patients with neurofibromatosis type 1. In aggregate, MPNSTs account for less than 5% of all soft tissue sarcomas.1 The lifetime risk of an MPNST in an individual with neurofibromatosis type 1 is 15% as measured in a Finnish population.2 Although neurofibromatosis type 1 is associated with MPNST, I will save discussion of that heritable condition for another post as to give it the appropriate degree of attention.
MPNSTs are thought to arise by a specific sequence of events that’s been defined over preceding decades. For a more in depth review, please see the this paper by Yao et al. 3 In essence, this happens through a two hit mechanism, or a loss of both copies of neurofibromin on chromosome 17. Loss of neurofibromin leads to less regulated growth, as neurofibromin inhibits action of the RAS pathway. The next steps in the process have multiple pathways, but most classically are CDKN2A (~63%) and PRC2 (70%).4

Unlike some other types of soft tissue sarcomas, there is not necessarily any stain that is pathognomonic for MPNST. A pathologist diagnoses morphologically, by anatomical considerations and by looking at some markers. S100 and SOX10 are only present in 50% of cases. The tumor may also be heterogeneous, incorporating muscle elements staining for common markers. Loss of H3K27me3 can also be a feature.5678
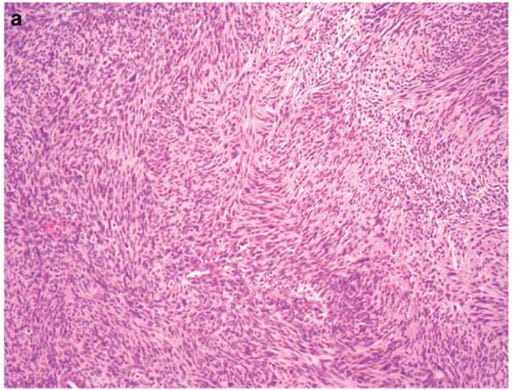
When a diagnosis is made, patients should have normal systemic staging in line with NCCN guidelines.
How is Malignant Peripheral Nerve Sheath Tumor Treated?
For patients with localized disease, wide local excision with negative margins is the mainstay of treatment. At diagnosis, less than 10% of MPNSTs are metastatic.9 The ability to remove tumors with wide margins can be complicated by the anatomy of MPNSTs given that when they arise from major nerves and removal of these nerves could cause significant morbidity. Prospective randomized trials of radiation for STS show improved local recurrent rates, but not overall survival. For larger MPNSTs that are high grade, radiation is a consideration for patients with MPNST, although our data are less precise and the role of RT merits further study. 10 There remains some concern regarding the long term side effects of radiation, and so careful consideration should be given to its administration.
For patients with advanced or metastatic disease, treatment is usually chemotherapy. Numerous attempts have been made to better define the optimal paradigms, nonetheless, most regimens rely on the knowledge base for soft tissue sarcomas in aggregate. Traditional chemotherapy is the first line treatment for most patients with unresectable disease. This is based on data that showed that the combination of Adriamycin and Ifosfamide was not superior from a survival standpoint for patients with STS.11

SARC006 was a small, phase 2 study conducted to evaluate the objective response rate by RECIST of patients with Stage III/IV, chemotherapy-naïve MPNSTs.12

This regimen utilized a combination of ifosfamide and doxorubicin followed by 2 cycles of ifosfamide/etoposide. 9 of 37 patients (sporadic and NF1 combined) had a partial response. 2 patients had progression. Unfortunately, the trial was unable to assess the endpoints for which it was designed. New patients were recruited slowly over the course of 6 years and ultimately it was closed due to poor enrollment. Later line regimens for patients with MPNST may incorporate other regimens approved for patients with STS such as gemcitabine/docetaxel or pazopanib.13 14
Unfortunately, multiple prior trials have failed to meet established endpoints. For example, SARC023, a trial of sirolimus, an mtor inhibitor, and ganetespib, an inhibitor of HSP90, did not meet criteria for clinical benefit. No responses were observed.15 SARC031 has also stopped recruitment.
Nonetheless, after failure of approved and active treatment, eligible and interested patients with MPNSTs should consider clinical trials. Here are a few ongoing studies at the time of writing:
ASTX727, a hypomethylating agent16
Dual checkpoint blockade (ipilimumab and nivolumab) in the neoadjuvant setting17
Clearly, the field has a great need for more, new, and effective therapies for our patients with MPNST.
Conclusions
MPNSTs are STSs that originate from nerve sheath cells. Approximately 50% of MPNSTs are a result of an inherited loss of neurofibromin (NF1). The majority of MPNSTs are high grade tumors. The diagnosis of MPNST may be difficult at the tumor can be heterogeneous and therefore interpretation of slides should be done at an expert center. Treatment of local disease, which is the majority of cases at presentation, relies on wide excision followed by radiation for patients with larger and higher grade tumors. Administration of systemic therapy as adjuvant treatment should be done on a case by case basis in consultation with an experienced oncologist. For patients with advanced disease, standard therapy has relied on cytotoxic chemotherapy such as doxorubicin based regimens. Ongoing trials seek to elucidate the efficacy of experimental treatments and should be considered when appropriate.
https://pubmed.ncbi.nlm.nih.gov/31081028/
https://www.mdpi.com/2072-6694/15/4/1077
https://academic.oup.com/noa/article/2/Supplement_1/i50/5645398
https://mp.uscap.org/article/S0893-3952(22)01951-2/fulltext
https://pubmed.ncbi.nlm.nih.gov/26645727/
https://publications.iarc.fr/Book-And-Report-Series/Who-Classification-Of-Tumours/Soft-Tissue-And-Bone-Tumours-2020
https://pubmed.ncbi.nlm.nih.gov/28752843/
https://pubmed.ncbi.nlm.nih.gov/26824706/
https://pubmed.ncbi.nlm.nih.gov/25452937/
https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(14)70063-4/fulltext
https://pubmed.ncbi.nlm.nih.gov/29138631/
https://pubmed.ncbi.nlm.nih.gov/28882536/
https://pubmed.ncbi.nlm.nih.gov/22595799/
https://pubmed.ncbi.nlm.nih.gov/32089640/
https://clinicaltrials.gov/ct2/show/NCT04872543
https://clinicaltrials.gov/ct2/show/NCT04465643