


Obligatory—This is not medical advice
When we discuss the many diagnoses that fall within the term sarcoma, some are closer to the mean than others. For instance, most sarcomas do not spread to lymph nodes, but there are exceptions. Clear Cell Sarcoma (CCS) is one of them. CCS, while commonly possessing an EWSR1 translocation is not as sensitive to cytotoxic treatment as other types of sarcoma, such as Ewing sarcoma for which EWSR1 is named. CCS exists in unique territory. Its historic name was melanoma of soft parts, and it can have melanocytic differentiation and pigment in 50% of cases. However, unlike melanoma, which bears a high response rate to immunotherapy1, CCS has not shown such a strong signal. 2 We continue to study and better understand the molecular and other mechanisms underlying CCS, which will be discussed below.
Epidemiology and Diagnosis of Clear Cell Sarcoma
Clear Cell Sarcoma (CCS) is an ultra-rare type of soft tissue sarcoma. In a review of the National Cancer Database, 489 patients were diagnosed with CCS in the United States between 2004 and 2014.3 The median age at diagnosis was 39 years. Common sites were the lower limb and hip. Most tumors were small by sarcoma standards with only a relatively low fraction of patients having metastatic, or stage IV, disease at the time of diagnosis.
When looked at underneath the microscope, CCS has spindled cells in groups (called nests).4 CCS can appear similar to melanoma and, as a result, the two are often in the differential of melanocytic neoplasms. Tumor cells will stain for for S-100, HMB45, Melan-A. They also can have pigment. The key distinguishing feature, however, is that CCSs have a translocation involving the EWSR1 gene on chromosome 22. In approximately 90% of cases, this is a fusion protein with the activating transcription factor-1 (ATF1) gene on chromosome 12. Oftentimes, it is only after genetic sequencing or fluorescent in situ hybridization (FISH) that a true diagnosis is obtained.

During the diagnostic window, imaging is also performed and careful attention should be paid to regional lymph nodes. There is some debate about the role of imaging of the brain (MRI), so this can be institution dependent.
Review of the national cancer database shows a correlation, as with other types of soft tissue sarcoma, between initial stage and survival (see below).



Treatment of Clear Cell Sarcoma
As with other varieties of sarcoma, treatment of localized disease involves resection of visible disease with consideration of adjuvant radiation depending upon marginal status and size. There is rarely thought to be a role for neoadjuvant or adjuvant cytotoxic treatment given the sensitivity of this entity to chemotherapy is low and prospective data in this subtype are lacking. A careful discussion is needed.
In the advanced disease setting, one of the larger CCS specific trials, the Phase II CREATE study tested crizotinib for patients primarily in a MET+ population. Crizotinib, a TKI, blocks the MET pathway. Therefore, there was strong preclinical rationale for its application in patients with clear cell sarcoma who had MET overexpression.5 28 patients were eligible for treatment. 17 patients had stable disease, and one had a partial response. This study was designed with a primary endpoint of response rate with a threshold of 10%, which was not met.


The most common treatment-related adverse events were nausea, fatigue, vomiting, diarrhea, constipation, and blurred vision. There were some patients who had durable control of their disease, and so it is often considered for most patients with clear cell sarcoma.
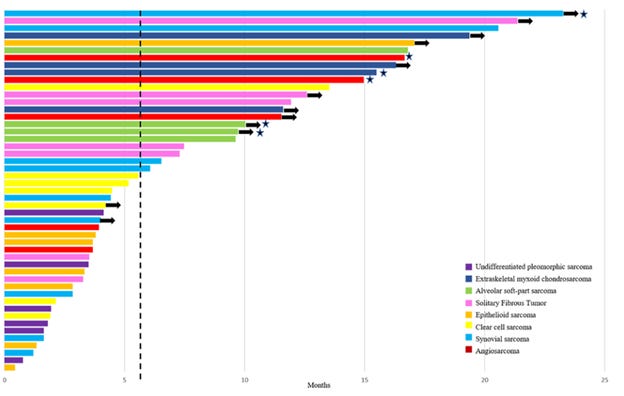
A review of patients treated between 1985 and May 2021 at 10 high volume sarcoma centers in 9 countries retrospectively evaluated efficacy of agents for patients. Their results are seen in the table below.

Unfortunately, in reviewing the results of that retrospective study, there were not many signals for high efficacy, although numbers were very small.
There, understandably, has been interest in exploring the role of immunotherapy in the treatment of patients with CCS, given its histologic similarities to melanoma. The phase IB portion of IMMUNOSARC had two partial responses to nivolumab and sunitinib, but these were short duration.6 While this is not enough data to make a firm recommendation, it may inspire future trials of combined TKI and immunotherapy.
Additional studies are needed for patients, and a few are underway. At the time of this writing, a Phase I/II open-label trial of Devimistat Plus Hydroxychloroquine is being conducted specifically for patients with clear cell sarcoma.7
Conclusions
The diagnosis of Clear Cell Sarcoma can be difficult and relies on both careful pathologic evaluation with stains as well as genetic features. Oftentimes this will require review at an academic center. Treatment of local disease relies on surgical management with excision and wide margins. Optimal therapy for advanced and metastatic disease is unknown, but studies have shown a possible role for tyrosine kinase inhibitors, particularly crizotinib for MET+ tumors. Immunotherapy has also been studied, but results have been mixed. Clear cell sarcoma is often less sensitive to cytotoxic therapy than other types of sarcoma, but it may merit consideration in some circumstances. Nuanced discussion is merited at each step in the treatment journey.
https://www.nejm.org/doi/full/10.1056/nejmoa1412082
https://www.esmoopen.com/action/showPdf?pii=S2059-7029%2822%2900143-0
https://pubmed.ncbi.nlm.nih.gov/19561568/
https://pubmed.ncbi.nlm.nih.gov/33203665/
https://clinicaltrials.gov/ct2/show/NCT04593758